Д.А. Охоботов, В.К. Карпов, А.А. Стригунов, О.Ю. Нестерова, Е.В. Афанасьевская, А.А. Камалов
Университетская клиника МНОИ МГУ им. Ломоносова, Москва
Молекулярные механизмы: основные медиаторы
1) Трансформирующий фактор роста- β1 (TGF-β1)
Важнейшим аспектом патогенеза болезни Пейрони является регуляция синтеза коллагена различными эндогенными и экзогенными факторами. В последние годы внимание исследователей привлек трансформирующий фактор роста-β1 (TGF‐β1), являющийся одним из наиболее фиброгенных цитокинов [1–3].
TGF-β1 синтезируется тромбоцитами, макрофагами, нейтрофилами и Т-лимфоцитами, эндотелиальными клетками, кератиноцитами и гладкомышечными клетками [4,5]. Он участвует в процессе воспаления, стимулирует синтез компонентов внеклеточного матрикса, регулирует пролиферацию и синтетическую активность фибробластов, а также их дифференцировку в миофибробласты, индуцирует образование активных форм кислорода, ингибирует синтез NO путем подавления активности индуцибельной NO-синтазы (iNOS), таким образом уменьшая соотношение NO/АФК [5–8]. Кроме того TGF-β1 рекрутирует из крови нейтрофилы, моноциты (увеличивая экспрессию MCP-1 – моноцитарный хемотаксический фактор-1), лимфоциты и фибробласты, ингибирует продукцию матриксных металлопротеаз (MMPs), особенно MMP-1, MMP-8, и MMP-13, которые обладают коллагенолитической активностью, увеличивает синтез ингибиторов матриксных металлопротеаз (TIMPs, особенно TIMP-1), увеличивает концентрацию MMP-10, вызывающей деградацию эластина [5,9]. TGF‐β1 активирует ядерный фактор каппа Б (NF-B), ингибирует фибринолиз путем увеличения продукции активатора ингибитора плазминогена (PAI-1) [5,7,10].
Существенную роль TGF‐β1 в патогенезе заболевания подтверждает ряд исследований. Так, например, в эксперименте на крысах, повторное введение в белочную оболочку аденовируса, экспрессирующего TGF‐β1, вызвало значительное искривление полового члена, что указывает на возможную роль TGF‐β1 в развитии болезни Пейрони [11]. Такое действие TGF‐β1 описывается, как «темная сторона» репарации тканей [9].
Роль TGF‐β1 подтверждают и эксперименты El-Sakka A.I. et al., проведенные в 1997 году на крысах. 18 крысам в белочную оболочку был введен цитомодуллин, который является синтетическим гаптапептидом с TGF-β-подобной активностью. Через 6 недель у 15 из 18 крыс наблюдалось истончение белочной оболочки полового члена с формированием бляшек, а также увеличенный уровень TGF-β1 mRNA и соответствующего белка. Кроме того, было отмечено отсутствие значительного увеличения двух других изоформ белка TGF-β2 и TGF-β3 [12]. Все это доказывает важную роль TGF-β1 в развитии заболевания, что в будущем может быть использовано для поиска новых терапевтических подходов к лечению больных болезнью Пейрони [13].
Повреждение белочной оболочки полового члена во время хирургического вмешательства с последующим восстановлением ее целостности приводит к возникновению гистологических изменений, схожих с таковыми во время острой фазы заболевания. Уже на первые сутки после операции почти у 85% крыс наблюдается повышение экспрессии TGF-β1, но на 3-й день повышенная концентрация цитокина наблюдается лишь у 50% особей, достигая 0 на 8-ой неделе. Тем не менее, хронизации процесса с появлением фиброзных бляшек не происходит, что может быть связано с качественным ушиванием раны и наличием адекватного гемостаза с минимизацией отложения фибриновых сгустков между оболочками полового члена. При микроповреждениях, наоборот, заживление происходит без какого-либо внешнего воздействия, что способствует дальнейшему прогрессированию заболевания [14].
2) Фактор роста тромбоцитов ( Platelet-derived growth factor – PDGF)
Тромбоцитарный фактор роста, наряду с TNF-α, TGF-β1 и FGF, является одним из профиброгенных медиаторов [2]. Он синтезируется, главным образом, тромбоцитами, в меньшей степени – макрофагами, проникшими в очаг воспаления. Как и TGF-β1, PDGF играет роль хемоаттрактанта для фибробластов, влияя на их синтетическую активность [5,15]. Кроме того, PDGF индуцирует образование TIMP-1 (ингибитор матриксной металлопротеазы-1), пролиферацию фибробластов и их дифференцировку в миофибробласты, а также рекрутирует остеобласты, что играет существенную роль в кальцификации и оссификации фиброзных бляшек [5,16].
3) Интерлейкин-1 (IL-1)
Интерлейкин-1 – провоспалительный цитокин, за синтез которого ответственны макрофаги, эндотелиальные клетки и фибробласты [4,5]. Для последних он является сильным хемоаттрактантом, одновременно стимулирующим образование MMP-1, MMP-2, MMP-8, MMP-9, MMP-10 и MMP-13, что, в свою очередь, способствует синтезу MMP-9 и MMP-3, а также bFGF (основной фактор роста фибробластов) [5,16,17]. Кроме того IL-1 активирует NF-κB-сигнальный путь, обеспечивающий дифференцировку и пролиферацию фибробластов, и индуцирует продукцию индуцибельной NO-синтазы (iNOS) [2,5,18].
Увеличение MMPs, как и их уменьшение может внести свой вклад в развитие фиброза путем изменения функций белков внеклеточного матрикса путем протеолиза, а также повышения концентрации факторов роста, таких как PDGF, VEGF, TGF-α, между элементами соединительной ткани [16].
4) Основной фактор роста фибробластов (bFGF)
О роли bFGF свидетельствует исследование Mulhall J.P. el al., проведенное в 2001 году, согласно которому синтез фактора роста фибробластов был в значительной степени повышен в клетках, взятых из бляшки пациента с болезнью Пейрони по сравнению с фибробластами нормальной ткани [19].
Основной фактор роста синтезируется фибробластами и миофибробластами, обеспечивая их пролиферацию [5,9,15]. Кроме того, он стимулирует синтез коллагена, увеличивает образование MMP-1 и MMP-9, TIMP-1 и способствует отложению фибриновых депозитов [5].
5) Ингибитор активатора плазминогена-1 (PAI-1)
Ингибитор активатора плазминогена-1, другой важный профибротический фактор, синтезируемый, главным образом, тромбоцитами [20]. Исследования показали, что экспрессия ингибитора активатора плазминогена-1 повышена в фиброзных бляшках у пациентов с болезнью Пейрони, что определяет его важную роль в патогенезе заболевания [21]. Он является физиологическим ингибитором двух главных активаторов плазминогена – тканевого типа (tPA) и урокиназного типа (uPA). PAI-1 необратимо связываясь с плазминогеном, образует комплекс, блокирующий действие tPA и uPA, тем самым препятствуя образованию плазмина и расщеплению сгустков фибрина [22].
Таким образом, PAI-1 выполняет роль стабилизатора фибрина в бляшке, предотвращая его деградацию и способствуя тем самым стимуляции фиброза [17,23]. Кроме того, он поддерживает развитие фиброзной индурации путем предотвращения распада коллагена ингибированием матриксных металлопротеиназ (MMPs) [5,23].
6) Фактор некроза опухолей TNF-α
TNF-α синтезируется моноцитами, макрофагами, Th1- и Th2-лимфоцитами и тучными клетками [5,9,17]. Он присутствует в высоких концентрациях во время острой фазы развития заболевания, постепенно уменьшаясь в процессе хронизации [1,4]. Его функции заключаются в следующем: привлечение фибробластов в очаг воспаления и стимуляция их пролиферации, стимуляция продукции iNOS, индукция клеточного апоптоза, стимуляция продукции MMP-9 (эластазы), активация транскрипционного фактора NF-κB, индукция синтеза PAI-1, увеличение проницаемости сосудистого русла [2,5,16,17].
7) Фактор, стимулирующий остеобласти 1 или плеотропин (osteoblast-stimulating factor-1 – OSF-1, PTN)
В исследовании Magee T.R. et al. в 2002 году в ткани больных болезнью Пейрони были обнаружены участки оссификации с высоким уровнем mRNA OSF-1, что дало основание предполагать определенную роль данного медиатора в патогенезе этого заболевания [3].
OSF-1, также известный как плейотрофин, HBNF1 или PTN представляет собой гепарин-связывающий белок, который стимулирует митотическую активность фибробластов, нейронов, остеобластов и ряда других клеток. OSF-1 вносит свой вклад в процесс фиброза путем увеличения количества фибробластов и, возможно, участвует в кальцификации бляшек путем привлечения в очаг воспаления остеобластов [3,7,8,15,16,24].
При болезни Пейрони наблюдается повышение концентрации профибротических факторов, вызывающих отложение коллагена и компонентов внеклеточного матрикса, что ведет к чрезмерной дезорганизации компонентов соединительной ткани [5]. В фиброзной бляшке присутствуют в основном коллагены двух типов – коллаген I и III, однако преобладает последний [5,6,25]. Волокна в бляшке беспорядочно ориентированы, отмечается уменьшение эластических волокон. Все это ведет к потере эластичности и неизбежному началу деформации полового члена [5].
Аутоиммунные нарушения
В качестве возможной причины, приводящей к возникновению болезни Пейрони, рассматриваются иммунологические механизмы с определенным вкладом аутоиммунного компонента [26–28]. Vande Berg J.S. et al. было выдвинуто предположение, согласно которому возникновение фиброзных бляшек представляет собой аутоиммунный ответ на повреждение сосудов ареолярной области [26].
Stewart S. et al. для подтверждения роли аутоиммунных патологических состояний в патогенезе болезни Пейрони исследовали спектр аутоантител к коллагену 1 и 3 типа, тропоэластину и α-эластину [27]. Эластин представляет собой белок, являющийся одним из важнейших компонентов внеклеточного матрикса. Он содержится в коже, кровеносных сосудах, легких, соединительной ткани, выполняя там свою основную функцию – обеспечение восстановления исходных размеров после растяжения, то есть эластичность [29]. Тропоэластин представляет собой мономерные молекулы, которые соединяются между собой двумя аминокислотами, образуя эластин [30]. α-эластин, наоборот, является продуктом деградации. Результаты оказались следующими: сывороточные аутоантитела к коллагену 1 и 3 типа были в пределах нормы, а антиэластиновые антитела была повышены у пациентов с болезнью Пейрони по сравнению с контрольной группой, что может указывать на вовлечение аутоиммунных механизмов в патогенез заболевания [27].
Позднее Ralph D. J. et al. изучили иммунологические особенности тканей, взятых у пациентов с фибропластической индурацией, что стало своеобразной поддержкой аутоиммунной теории болезни. Было выявлено наличие циркулирующих антинуклеарных антител, гипергаммаглобулинемия, отложение IgM антител в фиброзной бляшке и активация клеточного звена иммунитета, что подтверждается увеличением экспрессии человеческого лимфоцитарного антигена 2 класс (HLA II) на макрофагах. Однако антиген, вызывающий воспалительную реакцию и образование аутоантител не был обнаружен. Данные результаты позволяют предположить, что если основным этиологическим фактором болезни Пейрони является травма, то антиген, вероятно, входит в состав ткани полового члена, так как в ответ на повреждение происходит гиперактивация иммунитета с чрезмерной воспалительной реакцией и аномальным заживлением [28].
Дальнейшее развитие представлений о роли аутоиммунных патологических изменений нашли отражение в исследовании Schiavino D. et al., где пациентам был проведен ряд иммунологических тестов, характеризующих функционирование клеточно-опосредованного и гуморального иммунитета при болезни Пейрони. В результате установлено, что у 75,8% больных наблюдался, как минимум, один положительный тест по сравнению с 10% в контрольной группе. Нарушения в сфере клеточного иммунитета было у 48,5% пациентов, гуморального – у 31,8%, а аутоантитела (анти-ДНК, антинуклеарные антитела, антитела к гладкомышечным клеткам, к С3 и С4 компонентам комплимента) зафиксированы у 37,9%. Антинуклеарные и антигладкомышечные антитела выявлялись совместно у 7 (10,6%) пациентов [31]. Полученные данные еще раз подтверждают результаты предыдущих исследований, устанавливая определенную связь между патогенезом болезни Пейрони и иммунными нарушениями, однако точный механизм такой взаимосвязи все еще неизвестен.
Свободные радикалы и окислительный стресс
За последние 15 лет оксидативный стресс стал рассматриваться как один из ключевых моментов в патогенезе болезни Пейрони [15]. В месте воспаления, помимо выделения цитокинов, наблюдается гиперпродукция активных форм кислорода (АФК) [32].
Процесс оксидативного стресса начинается в течение 24–48 часов с момента травмы и является своеобразным триггером заболевания на первой ее стадии [33]. После активации клеток в очаге воспаления, происходит дегрануляция нейтрофилов с высвобождением большого количества лизосомальных ферментов, одновременно возникает своеобразный «респираторный взрыв», заключающийся в быстром высвобождении АФК (в частности, супероксид-радикалов и пероксида водорода) нейтрофилами, макрофагами, эндотелиальными клетками и тромбоцитами [34]. Механизм активации поддерживает внутриклеточный фермент никотинамидадениндинуклеотид фосфат (НАДФН), присутствующий в нейтрофилах, эозинофилах и макрофагах. НАДФН-оксидаза катализирует перенос электронов на молекулярный кислород, образуя супероксид-анион (O2•−). Он может давать начало ряду других АФК, таких как пероксид водорода, гидроксильный радикал, пероксинитрит и т.д. (см. таблицу) [5].
Таблица 1. Активные формы азота и кислорода
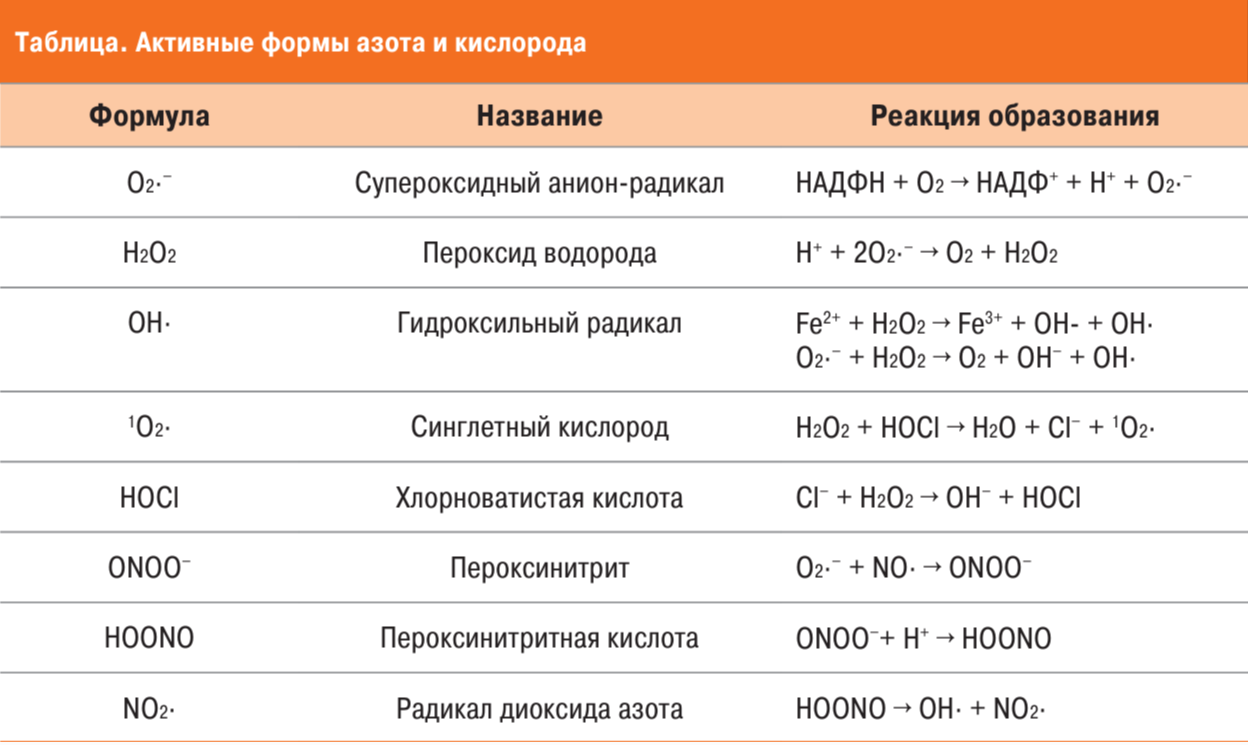
Большое выделение провоспалительных цитокинов и гиперпродукция АФК вызывают активацию транскрипционного фактора NF-kB, и дальнейшую продукцию воспалительных цитокинов c пролиферацией фибробластов [33]. Кроме того, NF-kB при болезни Пейрони он регулирует экспрессию генов, кодирующих FGF, TGF-β1, iNOS, коллагена, фибрина [35]. Индукция регуляции, iNOS приводит к повышению локального уровня оксида азота и ряда его метаболитов, называемых реактивными формами азота (RNS).
В норме NO основной медиатор эрекции, вызывающий расслабление гладких мышц пещеристых тел полового члена, а также участвующий в распаде коллагена путем активации MMPs, нейтрализации АФК, подавлении дифференцировки фибробластов [36]. В обычных условиях, эрекция обеспечивается за счет действия NO-синтазы, что является Ca2+ и кальмодуллин зависимым процессом. При болезни Пейрони, как было сказано выше, задействована индуцибельная NO-синтаза (iNOS, Ca2+ и кальмодуллин независимая), которая активируется провоспалительными цитокинами и вызывает быстрое локальное повышение концентрации NO. Оксид азота, в свою очередь, взаимодействует с супероксидом, образуя пероксинитрит. Пероксинитрит способен индуцировать апоптоз в эндотелиальных клетках, что в последующем приводит к снижению уровня свободного NO, повы шению адгезии тромбоцитов и лейкоцитов к эндотелию с высвобождением тромбоксана А2 и лейкотриенов. Все это ведет к вазоконстрикции с развитием эндотелиальной, а в дальнейшем и эректильной дисфункции [34].
Таким образом, при болезни Пейрони, как и при развитии ряда других заболеваний, таких как фиброз сердца и почек, аномальное заживление повреждений кожи, наблюдается нарушение соотношения свободного NO и АФК, а также других про- и антифибротических механимов. Это вносит свой вклад в патогенез заболевания, приводя к его усугублению и дальнейшему прогрессированию [8].
Заключение
Таким образом, основными патогенетическими механизмами развитиями болезни Пейрони являются аутоиммунные процессы, нарушение регуляции апоптоза, развитие оксидативного стресса, которые, действуя синергично, приводят к чрезмерной активации фибробластов с дальнейшим формированием фиброзной бляшки и искривлением полового члена различной степени.
Проведенный нами обзор литературы демонстрирует многообразие механизмов, участвующих в появлении фиброзных изменений полового члена. Каждый из них вносит определенный вклад в формирование бляшки, однако, единой концепции, объясняющей ее появление до сих пор нет. Считается, что триггерным фактором, запускающим острую фазу воспалительной реакции, являются возникающие во время полового акта микротравмы. Тем не менее, множество вопросов патогенеза болезни Пейрони, до сих пор остаются предметом дискуссии. Например, почему у одних пациентов заживление протекает без дальнейших анатомических изменений структуры полового члена, а у других воспаление переходит в хроническую стадию с формированием фиброзной бляшки. Возможно, путь репарации зависит от количества фибрина, который при достижении определенной критической концентрации играет основополагающую роль в запуске воспалительного каскада, а на докритическом уровне удаляется без каких-либо повреждений соседних структур. Литературные данные, способные подтвердить или опровергнуть данное предположение, отсутствуют, что создает необходимость проведения испытаний на животных моделях.
Помимо этого, определенный вклад в возникновение заболевания могут вносить бактериальные агенты, которые были обнаружены в одном из исследуемых образцов ткани как в виде изолированных, так и в виде свободно лежащих образований. Возникающие при этом гистологические изменения связаны с процессами реорганизации интерстициального матрикса с наличием участков кальцификации, а также характерными особенностями фибробластов, миофибробластов и гладкомышечных клеток. Следует отметить, что эксперименты, описывающие подобные изменения, были проведены около 30–40 лет назад. В связи с этим возникает потребность в повторном исследовании структуры фиброзной бляшки, особенно подлежащих структур, на электронно-микроскопическом уровне с использованием современных технологий для создания единой модели изучения этиологии и патогенеза заболевания, а также для единого стандартизированного описания гистологических изменений.
Для изучения ультраструктурных особенностей организации клеток, входящих в состав фиброзных бляшек, наиболее целесообразно их культивирование на различных питательных средах с оценкой максимальной плотности популяции и последующим микроскопическим исследованием сверхвысокого разрешения. Помимо этого клеточные культуры можно использовать как потенциальную модель для исследования химиотерапевтических методов лечения. Таким образом, патогенез болезни Пейрони многообразен, не имеет единой концепции и зависит от большого количества неучтенных факторов. Поэтому сегодня врач уролог лечит главным образом осложнение болезни Пейрони в виде анатомической деформации полового члена, а для уточнения механизмом возникновения фиброзных бляшек требуются дополнительные клинические и экспериментальные исследования.
ЛИТЕРАТУРА
- Zimmermann RP., Feil G., Bock C., Hoeltl L., Stenzl A. Significant alterations of serum cytokine levels in patients with Peyronie’s disease. International braz j urol : official journal of the Brazilian Society of Urology. 2008;34(4):457–66; discussion 466.
- Mulhall JP. Expanding the paradigm for plaque development in Peyronie’s disease. International journal of impotence research. 2003 Oct;15 Suppl 5:S93-102.
- Magee TR., Qian A., Rajfer J., Sander FC., Levine LA., Gonzalez-Cadavid NF. Gene expression profiles in the Peyronie’s disease plaque. Urology. 2002 Mar;59(3):451–7.
- Pavone C., Caruana G., Abbadessa D., Scaduto G., Gambino G., Serretta V., Alessandro R., Colomba P. Cytokine gene expression in the tunica albuginea of patients with Peyronie’s disease. Pilot study with a control group. Urologia. 2012 Jul;79(3):189–96.
- Paulis G., Romano G., Paulis L., Barletta D. Recent Pathophysiological Aspects of Peyronie’s Disease: Role of Free Radicals, Rationale, and Therapeutic Implications for Antioxidant Treatment-Literature Review. Advances in urology. 2017;2017:4653512.
- Gabbiani G. The myofibroblast in wound healing and fibrocontractive diseases. The Journal of pathology. 2003 Jul;200(4):500–3.
- Domes T., De Young L., O’Gorman DB., Gan BS., Bella AJ., Brock G. Is there a role for proteomics in Peyronie’s disease? The journal of sexual medicine. 2007 Jul;4(4 Pt 1):867–77.
- Gholami SS., Gonzalez-Cadavid NF., Lin C-S., Rajfer J., Lue TF. Peyronie’s disease: a review. The Journal of urology. 2003 Apr;169(4):1234–41.
- El-Sakka AI., Salabas E., Dincer M., Kadioglu A. The pathophysiology of Peyronie’s disease. Arab journal of urology. 2013 Sep;11(3):272–7.
- Szardening-Kirchner C., Konrad L., Hauck EW., Haag SM., Eickelberg O., Weidner W. Upregulation of mRNA expression of MCP-1 by TGF-beta1 in fibroblast cells from Peyronie’s disease. World journal of urology. 2009 Feb;27(1):123–30.
- Piao S., Ryu J-K., Shin H-Y., Zhang L., Song SU., Han J-Y., Park SH., Kim JM., Kim I-H., Kim S-J., Suh J-K. Repeated intratunical injection of adenovirus expressing transforming growth factor-beta1 in a rat induces penile curvature with tunical fibrotic plaque: a useful model for the study of Peyronie’s disease. International journal of andrology. 2008 Jun;31(3):346–53.
- El-Sakka AI., Hassoba HM., Chui RM., Bhatnagar RS., Dahiya R., Lue TF. An animal model of Peyronie’s-like condition associated with an increase of transforming growth factor beta mRNA and protein expression. The Journal of urology. 1997 Dec;158(6):2284–90.
- Haag SM., Hauck EW., Szardening-Kirchner C., Diemer T., Cha E-S., Weidner W., Eickelberg O. Alterations in the transforming growth factor (TGF)-beta pathway as a potential factor in the pathogenesis of Peyronie’s disease. European urology. 2007 Jan;51(1):255–61.
- El-Sakka AI., Selph CA., Yen TS., Dahiya R., Lue TF. The effect of surgical trauma on rat tunica albuginea. The Journal of urology. 1998 May;159(5):1700–7.
- Gonzalez-Cadavid NF., Magee TR., Ferrini M., Qian A., Vernet D., Rajfer J. Gene expression in Peyronie’s disease. International journal of impotence research. 2002 Oct;14(5):361–74.
- Herati AS., Pastuszak AW. The Genetic Basis of Peyronie Disease: A Review. Sexual medicine reviews. 2016 Jan;4(1):85–94.
- Campbell J., Alzubaidi R. Understanding the cellular basis and pathophysiology of Peyronie’s disease to optimize treatment for erectile dysfunction. Translational andrology and urology. 2017 Feb;6(1):46–59.
- Hayden MS., Ghosh S. NF-kappaB, the first quarter-century: remarkable progress and outstanding questions. Genes & development. 2012 Feb;26(3):203–34.
- Mulhall JP., Thom J., Lubrano T., Shankey T V. Basic fibroblast growth factor expression in Peyronie’s disease. The Journal of urology. 2001 Feb;165(2):419–23.
- Davila HH., Magee TR., Vernet D., Rajfer J., Gonzalez-Cadavid NF. Gene transfer of inducible nitric oxide synthase complementary DNA regresses the fibrotic plaque in an animal model of Peyronie’s disease. Biology of reproduction. 2004 Nov;71(5):1568–77.
- Davila HH., Magee TR., Zuniga FI., Rajfer J., Gonzalez-Cadavid NF. Peyronie’s disease associated with increase in plasminogen activator inhibitor in fibrotic plaque. Urology. 2005 Apr;65(4):645–8.
- Binder BR., Christ G., Gruber F., Grubic N., Hufnagl P., Krebs M., Mihaly J., Prager GW. Plasminogen activator inhibitor 1: physiological and pathophysiological roles. News in physiological sciences : an international journal of physiology produced jointly by the International Union of Physiological Sciences and the American Physiological Society. 2002 Apr;17:56–61.
- Gonzalez-Cadavid NF., Rajfer J. Mechanisms of Disease: new insights into the cellular and molecular pathology of Peyronie’s disease. Nature clinical practice Urology. 2005 Jun;2(6):291–7.
- Hauck EW., Hauptmann A., Haag SM., Weidner W. [New Insights into the Etiological Pathogenesis of Peyronie’s Disease]. Aktuelle Urologie. 2003 Oct;34(6):387–91.
- Hauck EW., Hauptmann A., Weidner W., Bein G., Hackstein H. Prospective analysis of HLA classes I and II antigen frequency in patients with Peyronie’s disease. The Journal of urology. 2003 Oct;170(4 Pt 1):1443–6.
- Vande Berg JS., Devine CJJ., Horton CE., Somers KD., Wright GLJ., Leffell MS., Dawson DM., Gleischman SH., Rowe MJ. Mechanisms of calcification in Peyronie’s disease. The Journal of urology. 1982 Jan;127(1):52–4.
- Stewart S., Malto M., Sandberg L., Colburn KK. Increased serum levels of anti-elastin antibodies in patients with Peyronie’s disease. The Journal of urology. 1994 Jul;152(1):105–6.
- Ralph DJ., Mirakian R., Pryor JP., Bottazzo GF. The immunological features of Peyronie’s disease. The Journal of urology. 1996 Jan;155(1):159–62.
- Weihermann AC., Lorencini M., Brohem CA., de Carvalho CM. Elastin structure and its involvement in skin photoageing. International journal of cosmetic science. 2017 Jun;39(3):241–7.
- Turino GM., Lin YY., He J., Cantor JO., Ma S. Elastin degradation: an effective biomarker in COPD. COPD. 2012 Aug;9(4):435–8.
- Schiavino D., Sasso F., Nucera E., Alcini E., Gulino G., Milani A., Patriarca G. Immunologic findings in Peyronie’s disease: a controlled study. Urology. 1997 Nov;50(5):764–8.
- Paulis G., Brancato T. Inflammatory mechanisms and oxidative stress in Peyronie’s disease: therapeutic “rationale” and related emerging treatment strategies. Inflammation & allergy drug targets. 2012 Feb;11(1):48–57.
- Sikka SC., Hellstrom WJG. Role of oxidative stress and antioxidants in Peyronie’s disease. International journal of impotence research. 2002 Oct;14(5):353–60.
- Agarwal A., Nandipati KC., Sharma RK., Zippe CD., Raina R. Role of oxidative stress in the pathophysiological mechanism of erectile dysfunction. Journal of andrology. 2006;27(3):335–47.
- Bivalacqua TJ., Champion HC., Hellstrom WJG. Implications of nitric oxide synthase isoforms in the pathophysiology of Peyronie’s disease. International journal of impotence research. 2002 Oct;14(5):345–52.
- Sasaki K., Hattori T., Fujisawa T., Takahashi K., Inoue H., Takigawa M. Nitric oxide mediates interleukin-1-induced gene expression of matrix metalloproteinases and basic fibroblast growth factor in cultured rabbit articular chondrocytes. Journal of biochemistry. 1998 Mar;123(3):431–9












Комментарии