Д.А. Охоботов, В.К. Карпов, А.А. Стригунов, О.Ю. Нестерова, Е.В. Афанасьевская, А.А. Камалов
Университетская клиника МНОИ МГУ им. Ломоносова, Москва
В настоящее время патофизиологические аспекты формирования фибропластической индурации полового члена (болезнь Пейрони) остаются не до конца понятными и вызывают ряд споров и противоречий. В данном обзоре проанализировано большинство существующих представлений о механизмах развития болезни Пейрони на клеточном и на молекулярном уровнях. Наиболее характерным аспектом развития фибропластической бляшки является чрезмерная активация фибробластов, которые под действием различных цитокинов усиленно синтезируют компоненты внеклеточного матрикса. К другим причинам можно отнести нарушение регуляции программируемой клеточной гибели, аутоиммунные процессы в организме, пагубное воздействие свободных радикалов. Однако точные механизмы, приводящие к возникновению вышеописанных нарушений, достоверно неизвестны. В связи с этим отмечается перспективность дальнейших исследований в этом направлении, так как более точное понимание патофизиологических аспектов болезни Пейрони позволит подобрать адекватную терапию и в будущем разработать эффективные методы профилактики, что сможет существенно улучшить качество жизни пациентов.
Фибропластическая индурация полового члена была впервые описана в 1743 году Франсуа Жиго де ла Пейрони, личным врачом короля Людовика XV. Однако, несмотря на значительный промежуток времени, исследователи мало продвинулись в понимании ее этиологии, патогенеза и, соответственно, эффективной профилактики и лечения [1–3]
Одной из наиболее известных теорий развития заболевания является теория аномальной фиброзной реакции заживления в ответ на микроповреждения, развивающиеся у генетически предрасположенных или иммунологически восприимчивых людей [2, 4, 5]. Своеобразным доказательством генетической предрасположенности является связь болезни Пейрони с другими заболеваниями с нарушением про- и антифибротических механизмов. Среди таких заболеваний контрактура Дюпюитрена, известная также как ладонный фиброматоз, болезнь Леддерхозе, или подошвенный фиброматоз [5–8]. Имеются данные о том, что для пациентов с вышеупомянутыми заболеваниями, как и для больных фибропластической индурацией полового члена, характерны изменения в генах, ответственных за синтез, деградацию и оссификацию коллагена, а также в генах, ответственных за дифференцировку миофибробластов [5–7].
Кроме того, есть ряд исследований, посвящённых ассоциации антигенов HLA II класса и болезни Пейрони [9, 10]. В работе Mazo E.B. et all. была отмечена связь фибропластической индурации с HLA-антигеном В8 [11]. В другом исследовании ученые выявили определённую связь заболевания с антигенами HLA-DQ5 [9]. Ralph D.J. et al. изучили ткани от 51 пациента, показав существенную корреляцию с HLA-B27 [12]. Тем не менее, существуют и противоречивые данные, поэтому вопрос о роли антигенов HLA остаётся открытым [13–15].
Таким образом, этиология и патогенез болезни Пейрони до конца не ясны, однако существует концепция о сочетании роли генетических факторов, как первичного звена, и травматического повреждения полового члена и иммунно-опосредованных механизмов в качестве вторичного компонента [8, 16, 17].
В настоящее время считается, что болезнь Пейрони – мультифакторное заболевание, развитие которого определяется комбинацией сразу нескольких элементов, которые будут рассмотрены ниже.
Особенности анатомического строения белочной оболочки и механизм формирования бляшек
Белочная оболочка плотно окутывает кавернозные тела полового члена и состоит из большого количества коллагеновых и эластических волокон, которые образуют толстые пучки, придающие половому члену жёсткость и эластичность [16]. Белочная оболочка лишена сосудов, а её кровоснабжение осуществляется за счёт артерий и вен так называемой ареолярной области, расположенной между ней и кавернозными телами [18]. Таким образом, при микротравмах, чаще возникающих во время полового акта, клетки воспалительного инфильтрата оказываются в своеобразной ловушке, формируя «пойманный в ловушку» воспалительный ответ [2, 3, 19].
Результатом такой воспалительной реакции является стимулируемое цитокинами лейкоцитов и макрофагов образование дополнительного внеклеточного матрикса. Цитокины не имеют возможности деградировать, накапливаются в замкнутом пространстве и стимулируют выработку ещё большего количества цитокинов, что приводит к синтезу коллагеновых волокон, фибронектина, протеогликанов [8, 20, 21]. Данный процесс рассматривается как своеобразная регенераторная реакция в ответ на микротравмы, целью которой является восстановить целостность белочной оболочки путём синтеза всё новых и новых коллагеновых волокон [2].
Заболевание имеет 3 стадии: субклиническая стадия, воспалительная стадия, стадия стабилизации. Первая стадия характеризуется коротким посттравматическим периодом около 2 недель, в течение которого накапливается фибрин и тромбоциты, происходит рекрутирование воспалительных клеток, макрофагов и лимфоцитов. Следующая стадия продолжительностью от 12 до 18 месяцев характеризуется продукцией факторов роста, вызывающих деградацию тканей с последующим синтезом нового внеклеточного матрикса. В течение этой стадии быстро появляются и растут фиброзные бляшки, что сопровождается болью и искривлением полового члена. С развитием воспалительной реакции происходит раздражение нервных окончаний, которые, как и сосуды, находятся в ареолярном слое, что вызывает боль при эрекции или без неё. В конце воспалительной стадии наблюдается стабилизация изменений, фиброкальцификация бляшек и гибель нервных волокон, в связи с чем исчезает болевой симптом. Формируется триада симптомов: бляшки, деформация полового члена и развитие эректильной дисфункции [16, 19, 22].
Как было сказано выше, триггерным фактором развития болезни является травма, после которой происходит повреждение капилляров микрососудистого русла, волокон ареолярной области и белочной оболочки, её деламинация с экстравазацией фибриногена [1, 2, 18]. В последующем он превращается в фибрин, накапливающийся между волокнами [8, 10, 23]. Фибрин посредством трансглутаминазы сшивается с фибронектином, что обеспечивают стабилизацию фибринового комплекса [4]. Этот комплекс действует как сильный хемотаксический фактор, рекрутирующий воспалительные клетки, такие как нейтрофильные гранулоциты, макрофаги, тучные клетки, фибробласты [4, 16]. Клетки немедленно начинают вырабатывать провоспалительные цитокины, в частности трансформирующий фактор роста β-1 (TGF-β1), активные формы кислорода, матриксные металлопротеазы, что способствует формированию фиброзной бляшки белочной оболочки [16, 21].
Основополагающая роль фибрина в запуске воспалительного каскада подтверждается исследованием Davila H.H. et al., проведённым в 2003 году. Установлено, что при введении в белочную оболочку полового члена крыс фибрина через 3 недели наблюдается возникновение фиброзной бляшки, подобной таковой при болезни Пейрони. Одновременно с этим наблюдается повышение уровня экспрессии TGF-β1, активных форм кислорода, а также дезорганизация эластических волокон (их укорочение или фрагментация), вызванная чрезмерной выработкой макрофагами фермента эластазы [24]. Продукты распада эластических волокон, в частности α-эластин, действуют как хемоаттрактанты, способствуя как увеличению воспалительного клеточного инфильтрата, так и притоку фибробластов [14]. Тромбоциты, уже присутствующие в области посттравматической экстравазации, также играют определённую роль в рассматриваемом процессе, высвобождая TGF-β1 и тромбоцитарный фактор роста (PDGF) [16].
Под действием вышеперечисленных факторов и ряда медиаторов, таких как PDGF, ИЛ-13, TNF-α, TGF-β1, ИЛ-1, FGF, активируются фибробласты с последующим синтезом компонентов внеклеточного матрикса, главным образом коллагена [2, 17, 19, 22]. В конечном итоге воспаление приобретает характер хронического процесса с чрезмерным накоплением и дезорганизацией в структуре коллагеновых и эластических волокон, что приводит к последующей кальцификации с образованием бляшек [7,22].
Исследование фиброзных бляшек на электронно-микроскопическим уровне показало наличие чрезмерной реорганизации интерстициального матрикса с нарушением периодичности коллагеновых волокон, формированием в некоторых случаях узелковых образований коллагена, а также признаками эластолиза и аномального эластогенеза с отложением свободно лежащего эластоподобного материала. Помимо этого отмечается наличие участков кальцификации, а также большое количество фибробластов, миофибробластов и гладкомышечных клеток с хорошо развитым синтетическим аппаратом, выростами клеточных мембран, охватывающих вышеописанные эластоподобные образования, а также большим количеством включений, представляющих собой микрофиламентный материал. Похожие электронно-плотные включения свободно располагаются между пучками коллагеновых волокон, что связывают отложением микрофиламентов в структурах интерстициальной ткани при разрушении клеток [25–27].
В образцах фиброзных бляшек с попавшей в срез эректильной тканью кавернозных тел, ядра некоторых гладкомышечных клеток и миофибробластов имеют звёздчатую форму и занимают большую часть цитоплазмы. Вокруг этих клеток часто наблюдается отложение микрофибрилл в виде аморфного эластиноподобного материала, что отсутствует в образцах контрольной группы [25].
Изменения, происходящие в интерстициальном матриксе, в некоторых случаях затрагивают и нервные окончания. Так, в исследовании Hirano D. et al. 1997 при проведении гистологического исследования фиброзных бляшек полового члена пациентов с болезнью Пейрони выявлена дегенерация миелиновых нервных волокон [26]. В другом исследовании, проведённом в 1981 году, у 4 пациентов из 20 отмечается полная фрагментация миелина нервных волокон, а нейротубулы и филаменты полностью отсутствуют. Цитоплазма шванновских клеток характеризуется наличием полосатых телец (зебра-подобные тельца), вакуолированных митохондрий и липидных включений. Помимо этого у 1 пациента в нескольких периваскулярных областях белочной оболочки наблюдается присутствие бактерий, которые на ультраструктурном уровне похожи на грамотрицательные. Эти микроорганизмы встречаются как в виде изолированных агрегатов, так и в виде свободно лежащих между коллагеновыми волокнами образований. Предполагается, что данные бактерии могут вносить вклад в дезорганизацию интерстициальной ткани и образование фиброзной бляшки путём выработки фермента гиалуронидазы [25].
Миофибробласты
Процесс образования фиброзной бляшки связан с активацией фибробластов и их последующей дифференцировкой в миофибробласты. Как первые, так и вторые ответственны за образование компонентов соединительной ткани у больных болезнью Пейрони, контрактурой Дюпюитрена, а также за процесс фиброза в целом [10, 28, 29].
Миофибробласты были открыты в 1976 году, а идентифицированы в бляшках пациентов с фибропластической индурацией полового члена позднее, в 1988 году. Вскоре стало понятно, что они играют ключевую роль практически во всех процессах, связанных с образованием компонентов соединительной ткани, как физиологического так и патологического характера [10, 30]. При заживлении ран и репарации миофибробласты секретируют коллаген и, сокращаясь, участвуют в уменьшении краёв раны, ускоряя таким образом заживление [4].
Считается, что миофибробласты образуются в результате дифференцировки фибробластов. Первичным стимулом является механическое давление, растяжение или травма, под действием которых фибробласт становится протомиофибробластом. Он характеризуется экспрессией цитоплазматических β- и γ-актина [30]. Затем под влиянием аутокринных и паракринных эффектов факторов роста, увеличивающихся при воспалительной реакции, в частности PDGF, ИЛ-13, TNF-α, TGF-β1, ИЛ-1, FGF протомиофибробласты превращаются в миофибробласты [2, 30, 31]. В процессе дифференцировки они приобретают промежуточный фенотип между их клетками-предшественниками и гладкомышечными клетками, экспрессируя α-гладкомышечный актин (α-SMA), который отсутствует в скелетных мышцах и фибробластах, и виментин, присутствующий у фибробластов, но нехарактерный для мышечных клеток [8, 28, 32].
Активированные миофибробласты секретируют цитокины, медиаторы воспаления, FGF, NO, активные формы кислорода, а также синтезируют матриксные белки, участвующие в репарации и образовании бляшки: коллагены, главным образом 1 и 3 типов, ламинины, протеогликаны [8, 10, 15, 30]. Синтетическая способность и сократительная функция активированных миофибробластов необходимы для заживления раны при нормальной репарации. После исполнения своей роли эти клетки должны элиминироваться путём апоптоза, тем не менее, в случае болезни Пейрони и контрактуры Дюпюитрена этого не происходит. Клетки персистируют, приводя в итоге к патологическому разрастанию соединительной ткани и фиброзу [8] (Рис. 1).
Участие фибробластов и миофибробластов в формировании фиброзной бляшки подтверждается исследованием Somers K.D. et al. в 1982 году. При культивировании клеток, полученных из бляшек пациентов с болезнью Пейрони, вырастает клеточная культура, состоящая из круглых и фибробластоподобных клеток часто с длинными цитоплазматическими выростами. Их изучение на электронно-микроскопическом уровне установило наличие звёздчатых ядер, а также цитоплазмы с большим количеством вакуолей. Последующий иммунофлуоресцентный анализ с антителами к гладкомышечному актину установил наличие интенсивного свечения по всей площади клетки и по контуру мембраны, что подтвердило их принадлежность к миофибробластам. В контрольной группе клеточной культуры наблюдался рост фибробластоподобных клеток без признаков миофибробластов. Данное исследование показало, что клетки фиброзных бляшек, формирующихся при фибропластической индурации полового члена, могут расти в условиях in vitro, что можно использовать как потенциальную модель для исследования химиотерапевтических методов лечения, а также более глубокого понимания патогенеза и этиологии заболевания [33].
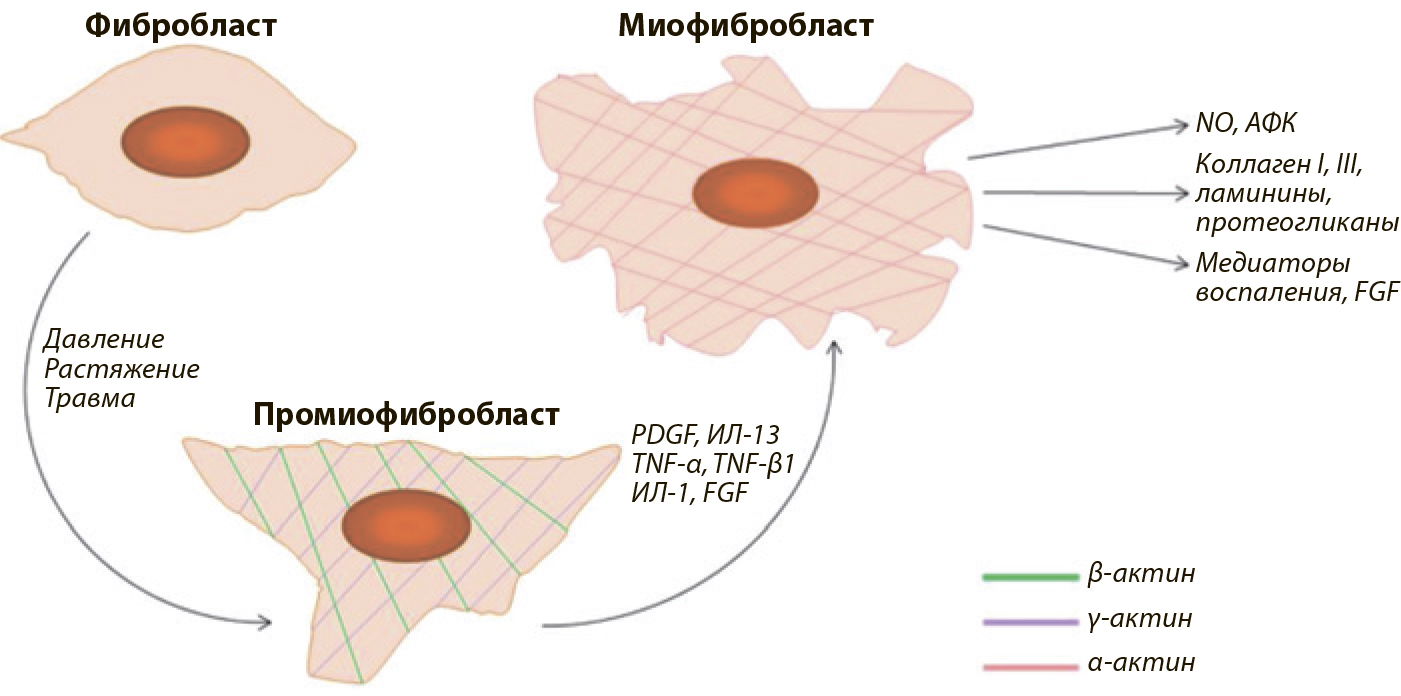
Рис.1. Модель дифференцировки фибробластов
Нарушение регуляции апоптоза
Апоптоз представляет собой регулируемый процесс программируемой клеточной гибели, в результате которого клетка распадается на отдельные апоптотические тельца, быстро фагоцитируемые макрофагами [34]. Одной из основных функций апоптоза является уничтожение дефектных, старых и выполнивших свою функцию клеток. Таким образом, обеспечивается поддержание клеточного гомеостаза, важные аспекты онтогенеза, регуляция пролиферации и дифференцировки [35, 36]. Кроме того, апоптоз является жизненно важным механизмом, участвующим в процессах нормальной репарации тканей и заживлении ран, элиминируя воспалительные клетки и способствуя превращению грануляционной ткани в рубцовую [37]. Дисрегуляция апоптоза в процессе заживления ран может привести к патологическим изменениям органа или его части, что будет проявляться в виде наличия чрезмерного рубцевания ткани или участка фиброза [36].
Как было сказано выше, формирование фиброзных бляшек белочной оболочки при фибропластической индурации полового члена рассматриваются многими авторами как результат аномального заживления микроповреждений, происходящих во время полового акта у предрасположенных людей [1, 4]. В процессе такой репарации при болезни Пейрони наблюдается гиперпролиферация фибробластов и миофибробластов с усиленным синтезом компонентов внеклеточного матрикса: коллагена, фибронектина, различных протеогликанов [3, 16].
Одним из возможных механизмов аномального заживления является нарушение регуляции программируемой клеточной гибели, что обеспечивает длительную персистенцию фибробластов, и, как следствие, приводит к фиброзным изменениям белочной оболочки полового члена [38]. Косвенным подтверждением данного утверждения может служить исследование, проведённое Watenabe M.S. et al. в 2017 году. Гистологический анализ ткани полового члена пациентов с болезнью Пейрони показал снижение уровня апоптоза клеток, по сравнению с контрольной группой, что может быть связано с нарушением молекулярных механизмов, обеспечивающих программируемую клеточную гибель [3].
В процессе клеточной гибели фибробластов и миофибробластов при болезни Пейрони, как было показано Loreto С. et al. в 2011 и 2014 годах, участвует как внутренний, так и внешний механизм апоптоза [38, 39]. В 2011 году Loreto С. et al. исследовали роль внешнего пути апоптоза у пациентов с фибропластической индурацией. В эксперименте изучили биопсию ткани белочной оболочки полового члена, взятую у 15 пациентов с болезнью Пейрони и у 4 пациентов без неё. Иммуногистохимическое исследование и Вестерн-блоттинг образцов выявили повышенный уровень TRAIL и DR5 у пациентов с болезнью Пейрони по сравнению с контрольной группой. Кроме того, гистологическая окраска гематоксилином-эозином позволила выявить атипичное расположение коллагеновых волокон в фиброзных бляшках с формированием скоплений, окружённых эластическими волокнами. При этом в нормальной ткани сохранялась нормальная структура внеклеточного матрикса: продольные волокна в наружном слое белочной оболочки, циркулярные – внутри [38].
В 2014 году была исследована активность внутреннего пути запуска апоптоза путём иммуногистохимической окраски образцов белочной оболочки, взятой у пациентов с болезнью Пейрони, на bax, bcl-2, каспазу 9 и каспазу 3. При этом отмечалась повышенная экспрессия bax, каспазы 3 и низкая – каспазы 9 и bcl-2 в фибробластах и миофибробластах. Полученные данные могут указывать на небольшой вклад внутреннего пути запуска апоптоза, по сравнению с внешним, в осуществлении процесса программируемой клеточной гибели у пациентов с болезнью Пейрони. Тем не менее, в связи с формированием фиброзных бляшек, их склонностью к стабилизации и наличием гиперпролиферации фибробластов с усиленным отложением внеклеточного матрикса гипотеза о возможном нарушении регуляции механизмов апоптоза остаётся актуальной [39].
В поисках подтверждения выдвинутой выше гипотезы группа исследователей Zorba O.U. et al. изучили уровни экспрессии генов различных апоптотических белков: FasR, FasL, Bcl-2, p53, каспазы 3 и 8 в бляшке и в неизменённой белочной оболочке у 8 пациентов. Было установлено, что FasR не экспрессируется ни в одном из образцов, экспрессия FasL в большей степени выражена в белочной оболочке, уровень Bcl-2 увеличен, а р53 повышен в бляшке. Кроме того, был обнаружен низкий уровень экспрессии каспазы 3 как в белочной оболочке, так и в бляшке. Экспрессия каспазы 8 была снижена во всех образцах, однако большее снижение наблюдалось в бляшке, по сравнению с нормальной белочной оболочкой. Р53 является транскрипционным фактором, ингибирующим клеточный цикл и индуцирующим внутренний путь запуска апоптоза. Увеличение Bcl-2 и уменьшение р53 может быть связано с высоким уровнем пролиферации и снижением клеточной гибели [40]. Полученные данные противоречат исследованию 2014 года, где уровень прокаспазы 3 был существенно повышен. Тем не менее, остальные результаты могут свидетельствовать в пользу ингибирования апоптоза миофибробластов и фибробластов, ведущего к стабилизации фиброзных изменений. Для подтверждения данного утверждения необходимо исследование на большей группе пациентов.
ЛИТЕРАТУРА
- Zimmermann RP., et al. Significant alterations of serum cytokine levels in patients with Peyronie’s disease. International braz j urol : official journal of the Brazilian Society of Urology. 2008;34(4):457–66; discussion 466.
- El-Sakka AI., et al. The pathophysiology of Peyronie’s disease. Arab journal of urology. 2013 Sep;11(3):272–7.
- Watanabe MS., et al. Mader AM., Nader HB., Glina S., Pinhal MAS. Extracellular matrix alterations in the Peyronie’s disease. Journal of advanced research. 2017 Jul;8(4):455–61.
- Gonzalez-Cadavid NF., Rajfer J. Mechanisms of Disease: new insights into the cellular and molecular pathology of Peyronie’s disease. Nature clinical practice Urology. 2005 Jun;2(6):291–7.
- Yafi FA., et al. Therapeutic advances in the treatment of Peyronie’s disease. Andrology. 2015 Jul;3(4):650–60.
- Herati AS., Pastuszak AW. The Genetic Basis of Peyronie Disease: A Review. Sexual medicine reviews. 2016 Jan;4(1):85–94.
- Capoccia E., Levine LA. Contemporary Review of Peyronie’s Disease Treatment. Current urology reports. 2018 May;19(7):51.
- Gonzalez-Cadavid NF., et al. Gene expression in Peyronie’s disease. International journal of impotence research. 2002 Oct;14(5):361–74.
- Nachtsheim DA., Rearden A. Peyronie’s disease is associated with an HLA class II antigen, HLA-DQ5, implying an autoimmune etiology. The Journal of urology. 1996 Oct;156(4):1330–4.
- Mulhall JP. Expanding the paradigm for plaque development in Peyronie’s disease. International journal of impotence research. 2003 Oct;15 Suppl 5:S93-102.
- Mazo EB., et al. Conservative treatment of Peyronie’s disease in the light of new pathogenetic data. Urologiia (Moscow, Russia : 1999). 2006;(2):32-35,37.
- Ralph DJ., et al. The genetic and bacteriological aspects of Peyronie’s disease. The Journal of urology. 1997 Jan;157(1):291–4.
- Leffell MS., et al.. Non-association of Peyronie’s disease with HLA B7 cross-reactive antigens. The Journal of urology. 1982 Jun;127(6):1223–4.
- Hauck EW., H et al. Prospective analysis of HLA classes I and II antigen frequency in patients with Peyronie’s disease. The Journal of urology. 2003 Oct;170(4 Pt 1):1443–6.
- Hauck EW., et al. New Insights into the Etiological Pathogenesis of Peyronie’s Disease. Aktuelle Urologie. 2003 Oct;34(6):387–91.
- Paulis G., R et al. Recent Pathophysiological Aspects of Peyronie’s Disease: Role of Free Radicals, Rationale, and Therapeutic Implications for Antioxidant Treatment-Literature Review. Advances in urology. 2017;2017:4653512.
- Szardening-Kirchner C., et al. Upregulation of mRNA expression of MCP-1 by TGF-beta1 in fibroblast cells from Peyronie’s disease. World journal of urology. 2009 Feb;27(1):123–30.
- Graziottin TM. The pathophysiology of Peyronie’s disease: beyond the Smith’s space. Vol. 41, International braz j urol : official journal of the Brazilian Society of Urology. Brazil; 2015. p. 1040–2.
- Gholami SS., et al. Peyronie’s disease: a review. The Journal of urology. 2003 Apr;169(4):1234–41.
- Davila HH., et al. Gene transfer of inducible nitric oxide synthase complementary DNA regresses the fibrotic plaque in an animal model of Peyronie’s disease. Biology of reproduction. 2004 Nov;71(5):1568–77.
- Magee TR., et al. Gene expression profiles in the Peyronie’s disease plaque. Urology. 2002 Mar;59(3):451–7.
- Campbell J., Alzubaidi R. Understanding the cellular basis and pathophysiology of Peyronie’s disease to optimize treatment for erectile dysfunction. Translational andrology and urology. 2017 Feb;6(1):46–59.
- Somers KD., Dawson DM. Fibrin deposition in Peyronie’s disease plaque. The Journal of urology. 1997 Jan;157(1):311–5.
- Davila HH., et al.Fibrin as an inducer of fibrosis in the tunica albuginea of the rat: a new animal model of Peyronie’s disease. BJU international. 2003 Jun;91(9):830–8.
- Vande Berg JS., et al. Peyronie’s disease: an electron microscopic study. The Journal of urology. 1981 Sep;126(3):333–6.
- Hirano D., et al. Electron microscopic study of the penile plaques and adjacent corpora cavernosa in Peyronie’s disease. International journal of urology : official journal of the Japanese Urological Association. 1997 May;4(3):274–8.
- Brock G., et al. The anatomy of the tunica albuginea in the normal penis and Peyronie’s disease. The Journal of urology. 1997 Jan;157(1):276–81.
- Gelbard M. Myofibroblasts and mechanotransduction: do forces in the tunica albuginea contribute to Peyronie’s disease? Vol. 5, The journal of sexual medicine. Netherlands; 2008. p. 2974–6.
- De Young LX., B et al. Protein biomarker analysis of primary Peyronie’s disease cells. The journal of sexual medicine. 2010 Jan;7(1 Pt 1):99–106.
- Gabbiani G. The myofibroblast in wound healing and fibrocontractive diseases. The Journal of pathology. 2003 Jul;200(4):500–3.
- Haag SM., et al. Alterations in the transforming growth factor (TGF)-beta pathway as a potential factor in the pathogenesis of Peyronie’s disease. European urology. 2007 Jan;51(1):255–61.
- Mulsow JJW., et al. Transforming growth factor-beta promotes pro-fibrotic behavior by serosal fibroblasts via PKC and ERK1/2 mitogen activated protein kinase cell signaling. Annals of surgery. 2005 Dec;242(6):880–7, discussion 887-9.
- Somers KD., et al. Cell culture of Peyronie’s disease plaque and normal penile tissue. The Journal of urology. 1982 Mar;127(3):585–8.
- Kaczanowski S. Apoptosis: its origin, history, maintenance and the medical implications for cancer and aging. Physical biology. 2016 May;13(3):31001.
- Monier B., Suzanne M. The Morphogenetic Role of Apoptosis. Current topics in developmental biology. 2015;114:335–62.
- Elmore S. Apoptosis: a review of programmed cell death. Toxicologic pathology. 2007 Jun;35(4):495–516.
- Greenhalgh DG. The role of apoptosis in wound healing. The international journal of biochemistry & cell biology. 1998 Sep;30(9):1019–30.
- Loreto C., et al. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and its death receptor (DR5) in Peyronie’s disease. A biomolecular study of apoptosis activation. The journal of sexual medicine. 2011 Jan;8(1):109–15.
- Loreto C., et al. The role of intrinsic pathway in apoptosis activation and progression in Peyronie’s disease. BioMed research international. 2014;2014:616149.
- Zorba OU., et al. Comparison of apoptotic gene expression profiles between Peyronie’s disease plaque and tunica albuginea. Advances in clinical and experimental medicine : official organ Wroclaw Medical University. 2012;21(5):607–14.












Комментарии